
Actinomycin D, which is active against several forms of cancer, has the distinction of being the first antibiotic discovered (in 1943) to possess useful antitumor activity. Because actinomycin acts essentially identically to block transcription in all cells, once it has gained access to the nucleus, it is equally applicable to the study of gene activity in eukaryotes as well as prokaryotes (it is generally ineffective against gram-negative bacteria simply because of a permeability problem, for if the walls of such cells are digested away, or otherwise weakened, the protoplast is revealed as fully sensitive to the antibiotic). A landmark was the determination of the structure of a crystalline 1:2 actinomycin:deoxyguanosine complex, which revealed for the first time that the antibiotic has an axis of near-perfect twofold rotational symmetry, which allows it to react with two guanine nucleosides in a symmetry-related fashion (6). With its tricyclic aromatic chromophore firmly embedded between the base pairs, the cyclic pentapeptide rings of the antibiotic are left neatly filling the minor groove of the distorted B-form helix, where they form numerous additional van der Waals contacts that help to stabilize the complex; positioned this way, their intrinsic right-hand twisted disposition with respect to the intercalated chromophore makes perfect sense, and the whole antibiotic molecule occupies a site covering about six base pairs in the minor groove. Detailed kinetic studies have revealed that both the association and dissociation reactions are complicated and require several rate constants to fit the data (3, 10). If the 2-amino group is removed from guanines (leaving inosinecytosine base pairs) and transferred to adenines (forming 2,6-diaminopurine-thymine base pairs), the sites to which actinomycin binds are relocated accordingly (11, 12). The molecular recognition process includes the formation of hydrogen bonds between the purine 2-amino groups and the carbonyl substituents of the L-threonine residues in the peptide rings of the antibiotic; there are also hydrogen bonds from the same threonine residues to the N(3) atoms of the purine nucleotides at the binding site (6, 12). Atherton (1977) In StructureActivity Relationships Among the Semisynthetic Antibiotics (D. Activation Energy the kinetic rates of chemical and enzyme-catalyzed reactions depend on temperature. The relationship for this dependence is known as the Arrhenius law: (1) where A and E are constants, R is the gas constant, and this the absolute temperature in Kelvin units. The activation energy E is the height of the energy barrier that the reaction must exceed to pass from reactants to products. Equation 1 can be expressed as: (2) Therefore the magnitude of the activation energy can be obtained from the slope of a plot of the log of a rate constant for a reaction as a function of 1/T. Many chemical and enzyme-catalyzed reactions increase the rate of reaction by two- to threefold for each 10°C increase in temperature. Although this relationship is useful in explaining the temperature dependence of reactions, it does not explain the rate in the thermodynamic terms of enthalpy H entropy S, or free energy G. Cornish-Bowden (1979) Fundamentals of Enzyme Kinetics, Butterworths, London, Boston, pp. Piszkiewicz (1977) Kinetics of Chemical and Enzyme-Catalyzed Reactions, Oxford University Press, New York, pp. Active Site the folding of a polypeptide chain that produces the final protein structure of an enzyme also leads to formation of the active site. From X-ray crystallography studies, it is apparent that the active site of an enzyme is a groove, cleft, or pocket that has access to the solvent and forms only a small part of the total solvent-accessible surface of the protein. The relatively large sizes of enzymes are undoubtedly due to the need to obtain, at the active site, the correct spatial relationships of the amino acid residues that are involved in binding of substrates, catalysis, and the release of products, as well as for any conformational changes associated with these steps. For substrates, these initial interactions are followed by the conformational changes that lead to the formation of the transition-state complex (see Transition State Analogue) and the chemistry for catalyzing the reaction brought about by reactive groups with the correct alignments. These may be the acidic, basic, and nucleophilic groups of the protein component of the enzyme, or the electrophilic groups of a prosthetic group (see Coenzyme, Cofactor). Active Site-Directed Irreversible Inhibitors Active site-directed irreversible inhibitors of enzymes are also known as active site-direcetd inactivating reagents, affinity labels, and photoaffinity labels. They combine the features of a substrate, or substrate analogue, with those of a group-specific reagent, as in affinity labeling, and have been used to determine the amino acid residues that are present in the active site and involved in enzymic catalysis. They are capable of binding specifically and reversibly at the active site of an enzyme and then causing inactivation through time-dependent covalent modification of an adjacent amino acid residue. The functional group of the inhibitor is usually an electrophile that can interact with an appropriately positioned nucleophile of the enzyme, to generate a covalent bond between them. Since these compounds are reactive in solution, they could also cause some nonspecific enzyme modifications. On the basis of this formulation, it would be expected that at the early stages of the interaction, I would behave as a competitive inhibitor with respect to the substrate. Photoaffinity labels, such as diazoketones and aryl azides, introduce a greater degree of specificity to the modification of amino acid residues, as they are not reactive in solution. It is only after the reversible interaction of the affinity label at the active site of an enzyme, and exposure of the resulting complex to light of the correct wavelength, that a highly reactive group is formed.
Metalloproteinases play an important role in tissue remodeling, in degenerative diseases such as rheumatoid arthritis, and in cancer. Several snake venom toxins are metalloproteinases, and a key enzyme in blood pressure regulation, angiotensin converting enzyme, is a metalloproteinase. A number of metal-binding substrate analogues have been found in nature (eg, phosphoramidon); and others, synthesized in the laboratory, are important pharmaceuticals. Metalloproteins Many proteins bind metal ions, permanently as prosthetic groups or more transiently as ligands. These metal ions play a variety of roles in metalloproteins: electron transfer, maintaining the protein structure, oxygen binding, forming coordinated hydroxide radicals, substrate binding, and electrophilic catalysis (see Metal-Requiring Enzymes and Metalloproteinases). During the past 15 years, it has become apparent that metalloproteins also play important roles in regulating the expression of genetic information. Minerals present in high concentrations, such as Na+, K+, and Mg2+, can play important roles in stabilizing nucleic acid structures, but they are unlikely to be widely used in gene regulation. Hence, it is not surprising that zinc, iron, and copper are prominent metalloregulators (see also CalciumBinding Proteins, Zinc-Binding Proteins, and Iron-Binding Proteins). The incorporation of a particular trace metal ion into an apoprotein is influenced by its ionic radius, thermodynamic stability, ligand-substitution kinetics, and charge (Table 1). Three to four ligands, usually the side chains of cysteine and histidine residues, typically complete the coordination sphere about a bound metal ion. Coordination Environments Preferred by Common Metal Ions Found in Proteins Metal Ion Coordination Number Geometry Zn2+ Fe2+ Cu+ Cd2+ 4 4 4 4 Tetrahedral Tetrahedral Tetrahedral Ligands His, Cys Cys Cys Tetrahedral His, Cys, Glu, Asp Hg2+ 3 Trigonal Cys Because of their intrinsic biological functions, regulatory metalloproteins are not normally present in large quantities in cell and tissue homogenates, and most are colorless or only weakly colored. Nonetheless, the study of regulatory metalloproteins has become the fastest growth area of inorganic biochemistry. The term "metalloregulation" refers to regulating the transfer of genetic information by metal ions. Regulatory metal-binding proteins are generally specific for a particular trace metal ion. For example, a synthetic zinc finger analogue (1) displays a strong preference for Zn2+ over Mn2+, Fe2+, Co2+, Ni2+, and Cd2+. Characterization As with any metalloprotein, the metal stoichiometry is of paramount importance. Several thousand putative metalloregulatory proteins have been reported in the literature, usually on the basis of limited sequence patterns alone. In the absence any physical characterization, however, such reports must be viewed as speculative because they offer no real evidence regarding the metal element involved. The nature of the metal coordination sphere can be probed by electronic absorption spectroscopy in cases involving iron or copper coordination. Zn2+ presents a challenge because it is colorless, but the Zn2+ can be replaced with Co2+, a chromophoric probe that possesses a comparable ionic radius, is kinetically labile, and prefers similar coordination environments. X-ray absorption spectroscopy is particularly useful for investigating the local environment (<~5) of the metals in these proteins (2). When X rays are absorbed by metal atoms, they liberate electrons that are backscattered from neighboring (ie, ligand) atoms. This interference phenomenon provides a very precise measurement of the distance to neighboring atoms. The edge region of the spectrum provides information about the valence state and coordination geometry of the metal and the identity of neighboring atoms. The structural methods do not address key issues regarding the way a regulatory protein acts as an information transducer. More precise structural information relating to the nature of the proteinnucleic acid contacts requires their cocrystallization and X-ray crystallographic structural determination. Approximately 50 structures of metalloproteins have been deposited to date in the Brookhaven Protein Data Bank (see Structure Databases). Regulatory Metalloproteins the major regulatory metalloproteins are described in Zinc-binding proteins, Zinc fingers, Ironbinding proteins, Iron-response elements, and Metal-response elements. In addition, organisms must frequently cope with toxic elements in the environment. Bacteria are especially rich sources of metalloregulators and encode resistance systems (3) for many toxic metal ions, including Hg2+, Tl+, Ag+, AsO2, Cd2+, and Cu2+.

It should be remembered, however, that such columns are of limited value, because they can be used only once. Because proteins are charged, they will detach from the column and migrate toward the appropriate electrode, if the column with the adsorbed M is exposed to a strong enough electric field. It was presumed that such an arm relieves the steric restrictions imposed by the backbone on the ligand, thereby increasing its flexibility and its availability to the protein (13). Initially, it was assumed that such hydrocarbon arms do not alter the inert nature of the matrix, a condition that obviously has to be ensured to preserve an active-site mediated adsorption of the extracted protein. This assumption seemed reasonable at the time because it had just been shown that at least some water-soluble proteins are quite well described as "an oil drop with a polar coat" (14), implying that the surface of water-soluble proteins is polar and thus not attracted to lipophilic "baits. The Limitations of Biospecificity-Interactions that are not Active Site-Mediated Proteins and their physiological ligands are multifunctional molecules whose functions involve a variety of physical interactions: hydrophobic, electrostatic, ion-dipole, and so on. Therefore, it is reasonable to assume that a protein might interact with a column coated with a ligand (very often anchored to the beads at a local concentration much higher than its concentration in vivo) not only by means of its active site. While it is sometimes possible to minimize these nonspecific interactions, it is not always possible to avoid such interfering effects, because they may be an intrinsic property of the system. Other proteins, in addition to the desired one, may therefore "regard" the column material as an ion exchanger by virtue of its triphosphate groups, or as a hydrophobic column by virtue of its adenine moieties. The efficiency of resolution will then depend on the magnitude of the affinity produced by chargecharge or hydrophobic interactions, as compared to the affinity between the active site of the desired macromolecule and its immobilized substrate or effector analogue. With columns of macromolecular ligands (eg, enzyme subunits, antibodies, lectins), the probability of encountering such built-in interfering effects is considerably higher, because their immobilization usually involves different anchoring points. This leads to a heterogeneous presentation of the various regions of the ligand macromolecule. In some of these presentations, the biospecific active site is available for interaction, while in other presentations the active site itself is inaccessible or sterically hindered. Hydrophobic patches in such ligands may be available for interaction not only in the biospecifically functional presentation, but also in other presentations. In fact, the tendency of a lectin such as concanavalin A to adsorb onto hydrophobic substances, in addition to its binding to sugars of the mannosyl configuration, was observed in several laboratories. However, common biorecognition elements may also be found with proteins having no apparent functional similarity. It seems, therefore, that the retardation of the free catalytic subunit on the immobilized inhibitor is due (at least in part) to an affinity between the inhibitor and recognition subsites at the active site of the enzyme. Affinity Electrophoresis By analogy to affinity chromatography, it is possible to introduce specific ligands for a macromolecule into the gels of gel electrophoresis and to measure the specific retardation of the macromolecule due to its interaction with such a reagent. The advantage of such affinity methods lies in the augmented resolving power conferred by the specificity of the binding interaction. In cross electrophoresis, a ligand with a net charge opposite to the species of interest migrates electrophoretically into the gel in the opposite direction. Macromolecular substrates within a gel may serve as immobilized affinity reagents, either by themselves or as carriers of covalently attached affinity groups. The magnitude of the electrophoretic retardation depends on the concentration of the affinity reagent in the gel; quantitative determination of this relationship makes it possible to estimate the apparent association constant for binding of the ligand to the sample. Further information concerning the interaction can be gained from affinity electrophoresis by variation of the buffer composition (eg, the addition of metal ions to the buffer), the pH, or the temperature. Affinity Labeling Affinity labeling is a strategy to modify chemically an amino acid residue within a specific ligandbinding site of an enzyme, either at the active site or at a regulatory, allosteric site. In this approach, a reagent is designed that resembles structurally the natural ligand of the enzyme but features in addition a functional group capable of reacting covalently and indiscriminately with many different amino acid residues. The designed reagent is intended to mimic the natural ligand in forming a reversible enzyme-reagent complex analogous to the enzyme-substrate complex and, once directed to that specific site, to react irreversibly with an amino acid residue accessible from that site. In the case of a purified enzyme, such a reagent allows identification of a particular ligand binding site or domain, which can be an experimental evaluation of a predicted binding site location based on recognition of a protein motif from its amino acid sequence. Affinity labeling constitutes a valuable starting point for selecting appropriate target sites for subsequent site-directed mutagenesis experiments and is an important tool in probing structurefunction relationships in enzymes.
In addition to having a biosynthetic pathway for protein secretion, many eukaryotic cells have an endocytosis pathway that internalizes plasma membrane components and returns most of them to the cell surface (4). This membrane recycling pathway allows a cell to move membrane to a site where it might be more needed; for example, a region of cell growth or migration (5). The recycling pathway also allows a cell to internalize nutrients bound to receptors, to remove the nutrients, and to send the receptors back to the cell surface for more. In the final step, a membrane vesicle on the recycling pathway must fuse with the plasma membrane (that is, it must undergo exocytosis). Thus, the exocytosis machinery is essential for endocytotic recycling, as well as for protein secretion. Exocytosis is used in other processes besides delivery of secretory proteins to the cell surface. As with protein secretion, there appear to be constitutive and regulated exocytotic steps associated with membrane recycling. Membrane vesicles carrying nutrient receptors, such as the receptors for transferrin and low-density lipoprotein, fuse rapidly with the plasma membrane, independently of an external signal. In specialized cases, exocytosis is inhibited, causing an accumulation of endocytotically derived membrane vesicles (6). Fortunately, similar molecular machinery seems to be used by all forms of exocytosis, regulated or constitutive, biosynthetic, or plasma-membrane recycling. Furthermore, the molecules that regulate exocytosis appear to be conserved, as are the transporters that put neurotransmitters into dense core secretory granules and into synaptic vesicles. Measurements of Exocytosis A common but crude measure of exocytosis is the appearance in the extracellular medium of the contents of a secretory vesicle that has undergone exocytosis. Thus exocytosis of synaptic vesicles (see Secretory Vesicles/Granules) at the nerve terminal causes release of neurotransmitter, and exocytosis of secretory granules from b-cells of the pancreatic islets of Langerhans releases insulin. Extracellular neurotransmitters and hormones can be measured biochemically, as can the constitutive secretion of enzymes such as invertase by yeast or proteinases by mammalian cells. Biochemical measurements are usually incapable of measuring the kinetics of exocytosis with accuracy. The release of neurotransmitters, however, can be measured electrophysiologically with much greater precision. Most neurotransmitters activate the postsynaptic target cell by binding to closed ion channels and causing them to open. Measurement of current flow through the postsynaptic ion channels gives an almost instantaneous assay of the exocytosis rates in the presynaptic nerve terminal. The precision and sensitivity of electrophysiological measurements, pioneered by Dr. Bernhard Katz and his coworkers at University College, London, allowed detection of a quantum of neurotransmitter release, lasting a millisecond or so, generated by exocytosis of the contents of a single synaptic vesicle. Electrophysiology revealed that exocytosis is very rapid, occurring within 100 µs of a stimulus to the nerve terminal, and that the concentration of the neurotransmitter in the synaptic vesicle is very high, greater than 100 mM. Postsynaptic currents generated by presynaptic release of neurotransmitter accurately measured the exocytosis because the receptor channels were 10 to 20 nm from the exocytosis site. Release of biogenic amines from cells such as chromaffin cells can likewise be measured rapidly and quantitatively by placing a carbon electrode close to the cell and detecting the biogenic amine by its redox potential (8). Another technique widely used to study exocytosis uses the cell-surface capacitance. The electrical capacitance across the plasma membrane of a cell is proportional to its area. When exocytosis occurs, the capacitance increases because of the addition of membrane (9, 10). The sensitivity of capacitance measurements is such that it can measure the addition of the membrane of a single secretory granule. It cannot yet detect the addition of a single synaptic vesicle, since they are too small (50 nm diameter). When the capacitance signal is big enough, however, the capacitance changes give an instantaneous measurement of exocytosis. To measure capacitance, a cell must be attached to a "patch electrode," which allows electrical communication between the salt solution inside the electrode and the cytoplasm of the cell.

All tumor cells that have been examined have been found to secrete Ang, and in at least one case, pancreatic cancer, the aggressiveness of the tumor is related to the amount of angiogenin produced (6). Ang secreted by tumor cells migrates through the extracellular matrix until it reaches an endothelial cell. Degradation of the extracellular matrix-for example, by plasmin-activated matrix metalloproteinases-may stimulate a few endothelial cells to migrate, and this could trigger expression of the Ang receptor. Binding of Ang to the receptor would then, on the one hand, initiate a second-messenger response, perhaps through activation of the above-mentioned phospholipases, and, on the other, promote endocytosis and nuclear localization of Ang. The ribonucleolytic action of Ang within the nucleolus would, together with signals generated via the second messengers, activate processes leading to cell proliferation. The proliferating endothelial cells would migrate through the now degraded extracellular matrix toward the tumor cell from which the Ang was released. The cell-adhesion properties of Ang may be important for ensuring cell migration in the proper direction. At present, this view of the mode of action of Ang is largely speculative, particularly because nothing is known about the events that occur within the nucleus and lead to cell division. Nevertheless, it summarizes current thinking and is a useful basis for further investigation. This being the case, anti-angiogenin agents could have potential importance in the treatment of cancer and other angiogenesis-related diseases. Similar results have been observed with other types of tumors, and when used in conjunction with conventional therapeutic agents, a synergistic effect was seen (2). Actin was also tested for its anti-tumor activity, and it too was capable of preventing tumor growth in more than 60% of treated animals. It should be noted that anti-angiogenin agents do not affect the growth of already-established tumors. They are only effective when administered to animals at the same time as the tumor cells. They are thought to act by specific extracellular inactivation of tumor-secreted Ang and the consequent inhibition of tumor angiogenesis. These experiments are important in that they provide clear evidence of a crucial role for angiogenin in the early stages of development of these tumors. Other Biological Properties One of the major complications associated with regular hemodialysis is the increased morbidity and mortality arising from infections. Indeed, a number of compounds have been isolated from uremic serum and shown to inhibit the biological activity of these white cells. One of these is an inhibitor of leukocyte degranulation that turns out to be Ang (7). Nanomolar concentrations of Ang inhibit both spontaneous and peptide-stimulated degranulation by 60% and 30%, respectively. However, Ang has no other effect on the cellular responses of polymorphonuclear leukocytes, such as chemotaxis, phagocytosis or their peptide-stimulated oxidative respiratory burst. Ang has also been reported to suppress significantly the proliferation of human lymphocytes stimulated by a mixed-lymphocyte culture or by phytohemagglutinin or concanavalin A (8). It was thought that this effect might synergize with the effect of Ang on neovascularization of tumors and thereby contribute to tumor development. Conclusions Ang is now recognized as a pleiotropic molecule capable of inducing several intra- and extracellular activities. Characterization of its cellular receptors, elucidation of its mechanism of nuclear translocation, identification of its intranucleolar target substrate, and understanding how these events result in cellular proliferation and blood vessel formation will provide important and novel information that can be utilized for either promoting or inhibiting angiogenesis for therapeutic purposes. Riordan (1997) "Structure and Function of Angiogenin" In Ribonucleases: Structures and Functions (G. Anhydrides "Anhydride" means "without water," and many anhydrides are present in nature. An acid anhydride [I] is formed by eliminating a molecule of water from two acids (Scheme 1): Adding a molecule of water reverses the reaction. Practically, a carboxylic acid anhydride is prepared by the nucleophilic displacement of chloride ion from acyl chlorides by carboxylate ion. They react with water to yield acids, with amino groups to yield amides, and with alcohols to yield esters. Acid anhydrides are utilized as reactive intermediates in organic syntheses, such as peptide synthesis, and sometimes as enzymesubstrate intermediates.
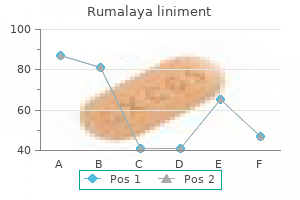
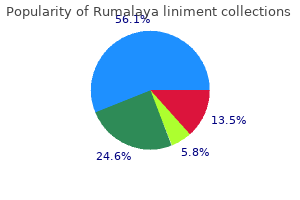
Cupric Sulfate (Copper). Rumalaya liniment.
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96868
Normal cells plated on a confluent monolayer will give a varying response, depending on the cell lineage of the confluent monolayer. If the plated normal cells are of the same lineage as the confluent monolayer, they will not grow; for instance, fibroblasts will not grow on a confluent monolayer of fibroblasts, and normal glial cells will not grow on a confluent monolayer of normal glial cells (15), whereas their transformed counterparts will. On the other hand, normal glial cells will grow on a confluent monolayer of fibroblasts, and normal keratinocytes will grow on a monolayer of normal 3T3 cells (16). Cultures that are propagated in suspension will, of course, not encounter contact inhibition. Part of this is undoubtedly due to nutrient depletion, catabolite buildup, and pH depression, but even if limiting nutrients are replaced and the pH is stabilized, high density suspension cultures do not increase significantly above 12 Ч 106 cells/ml. Quite often suspension cultures, such as hybridomas, will remain in the plateau phase for only a short time and then deteriorate rapidly, leaving few viable cells in the culture. The reason for this appears to be that the cells tend to enter apoptosis when they reach a high density. Some moderate success has been achieved overexpressing bcl in these cells to inhibit apoptosis (17). Freshney (1994) Culture of Animal Cells, a Manual of Basic Technique, Wiley-Liss, New York, pp. Contact Maps Contact maps are two-dimensional representations of three-dimensional protein structures. A threedimensional description of a protein structure composed of N structural units could be expressed as an N Ч N array of the pairwise distances (see Distance Geometry). This could be done for all pairs of atoms, for selected types of atoms (eg, Ca atoms), for groups of atoms (eg, side-chain centers of mass), or for entire amino-acid residues. Contact maps are generated from such matrices by taking a certain cutoff value for the pairwise distances. For example, the N Ч N matrix of the distances between protein Ca atoms (1, 2) can be transformed into a Ca-based contact map (3). Those Ca atoms that are closer to each other in the protein structure than the chosen cutoff distance are considered to be "in contact. Alternatively, one may assume a set of several critical values for the distances between Ca atoms (or for other atoms or groups of atoms) and generate an integer matrix-the equivalent of a contact map with a colored or gray scale. A map of main-chain hydrogen bonds could also be considered as a variant of a protein contact map. The choice of structural units being mapped and the choice of cutoff distances determine the quality and range of structural information being stored in a contact map (see. Schematic drawing of the structure of the B domain of protein G and its contact maps. The amino acid sequence of the protein is given along each axis in one-letter code, and the secondary structure is indicated as follows: E is extended b-strand, H is helix, and S and T are two types of turns. Above the diagonal, the dark squares correspond to the pairs of side chains for which the distance between at least one pair of heavy atoms is less than 5 Е. The amino acid sequence of the protein is given along each axis in oneletter code, and the secondary structure is indicated as follows: E is extended b-strand, H is helix, and S and T are two types of turns. Contact Maps as a Fingerprint of Protein Three-Dimensional Structure A contact map constitutes a structural "fingerprint" of a protein (4). The secondary structure, fold topology, and side-chain packing patterns (for side-chain contact maps) can be visualized conveniently and read from the contact map. Furthermore, structural similarity between a pair of proteins is immediately apparent by a very pronounced similarity of their contact maps; in comparing two protein structures, there is no need to search all their possible relative orientations. The reconstruction of a protein structure from its contact map is more complex, although low-to-moderate resolution three-dimensional models can be easily built, even from a fragmentary contact map (5). The accuracy of the model depends on the type of contact map and the computational tools employed. Ca-Based Contact Maps Ca-based contact maps and distance matrices were perhaps the first commonly used maps for visualization of protein structures (3, 8, 9).
Syndromes
Finally, in the case of the Escherichia coli tryptophanase operon, it appears that ribosomes are used as the regulatory molecule. In addition, several other systems function similarly but appear to have arisen independently. A general model for antitermination control by the Sac/Bgl family of antiterminator proteins. Under noninducing conditions, transcription starts at the promoter (designated by the arrow) and terminates prematurely, often in a leader region prior to the structural genes. The operon is cryptic in wild-type strains but can become functional through spontaneous mutations. When functional, expression of this operon is regulated by antitermination mediated by the BglG protein in response to the levels of b-glucosides (4). Phosphorylation of both b-glucosides and BglG is accomplished by transfer of the phosphate group from the same phosphorylated residue, Cys24, in BglF (6). These results suggest that, under conditions in which b-glucoside levels are high, the phosphate group can be transferred from BglG back to Cys24 in BglF. A model has been proposed in which unliganded BglF phosphorylates BglG, and b-glucoside binding induces BglF to undergo a conformational change that activates it to dephosphorylate BglG. A similar system for b-glucoside utilization exists in the related Gram-negative enteric bacterium Erwinia chrystanthemi, although in this case the arb operon is not cryptic (7). ArbG shows high sequence similarity to BglG, suggesting that it functions analogously in antitermination control of the E. Antitermination also appears to control b-glucoside operons in several Gram-Positive Bacteria as well. In addition, a protein, BglR, with homology to BglG also controls b-glucoside usage in Lactococcus lactis (9). SacT and SacY show extensive sequence similarity to each other, as well as to BglG from E. The antitermination mechanisms that control these genes also appear to be quite similar to that described above for the E. The domain exists as a dimer, with each monomer consisting of a four-stranded antiparallel beta-sheet. These residues are clustered on the surface of one side of the protein structure (15). In Lactobacillus casei, the lactose (lac) operon is regulated in response to lactose levels by LacT, which shows sequence homology to the other members of the Bgl/Sac family of antiterminators (17). The amino acid sequences of these antiterminator proteins are not similar to any other antiterminator proteins. The amidase (ami) operon of Pseudomonas aeruginosa is regulated by antitermination in response to short-chain aliphatic amides, such as acetamide. The amiR gene encodes an antiterminator protein (AmiR), which is negatively regulated by AmiC, apparently through formation of an AmiC-AmiR complex (20). Acetamide destabilizes the AmiC-AmiR complex, leading to antitermination and expression of the operon. AmiR binding has been suggested to function in antitermination by interfering directly with formation of the terminator stem-loop structure (20). In addition to all the catabolic operons described above, one anabolic operon has been shown to be regulated by antitermination. Expression of the nas operon of Klebsiella pneumoniae, which encodes enzymes required for nitrate assimilation in this bacterium, is induced by nitrate or nitrite. The NasR protein mediates transcription antitermination through a terminator in the leader region of the operon (21). Expression of these genes is induced specifically by starvation for the corresponding amino acid. In the case of the amino acid operons, insufficient levels of the amino acid leads to increased expression of the corresponding biosynthetic operon. Hence, in the absence of the inducing signal, transcription terminates prematurely in the leader region prior to the coding sequences. In addition to the conserved secondary structures, there is an important conserved 14-nucleotide sequence known as the T-box present in each leader region; hence these genes are known as the T-box family. An alternate arrangement of the leader region involving base-pairing between a portion of the T-box and a conserved sequence in the 5 side of the terminator stem has been proposed to form an antiterminator structure that allows transcription to read through into the structural genes.
Over time, a pellet of sedimented material is deposited along the most radially distant wall of the tube containing the sample (Table 3). At a sufficiently high rotor speed (and therefore high centrifugal force), virtually all macromolecular solute components heavier than the solvent are pelleted out of solution over time. Because it is possible to select a variety of rotor velocities, sample tube dimensions, rotor types, centrifugation run times, etc. For example, large components are typically removed by sedimentation at low speeds for short periods of time. If the supernatant from such a centrifugation is placed in a new container and centrifuged again at a higher rotor velocity for an appropriate period of time, it is possible to collect another distribution of particles from solution, this time of smaller dimensions than the first. Therefore repeating this process yields a series of pelleted samples with distributions of solute molecules, each of which have accumulated under conditions differing only in their centrifugal forces and thus have different sedimentation rates. Most samples of biological interest are complex mixtures of interacting solutes in solution at widely different concentrations. Because the size, shape, and concentration distributions of these components are routinely broad, together they exhibit very broad distributions of sedimentation rates. Thus the technique of solute pelleting, though widely applied, is only rarely sufficient to purify a single component completely. Typically it is used to selectively enrich, often to a high degree, a preparation of cellular or subcellular components in a single component. For small biological solutes, such as soluble cytosolic proteins prepared by a protein isolation procedure, pelleting is routinely employed along with differential precipitation (salting out) using ammonium sulfate (see Sulfate Salts) to yield highly enriched, sedimented precipitates of the desired protein components. Square-root speed Used reduction to calculate the permissible rotor speed in rpm when using law nonprecipitating gradient materials at densities greater than the manufacturer permits in a rotor Pelleting is also of great use when applied to harvesting viruses or cells from various growth media. When cells or viruses are used to produce various recombinant proteins of pharmaceutical interest, centrifugal harvesting is often employed to capture the biological component from the (usually large) fermentation broth. In these applications, a preparative centrifuge and rotor capable of accepting a continuous flow into and through the rotor are employed. The centrifugal force applied to the sample stream as it passes through the rotor is adjusted to permit selective pelleting of cells within the centrifuge rotor. The cell-depleted broth passes out of the rotor/centrifuge and into a receiver vessel, and, then the harvested cells are recovered from the centrifuge rotor. A short, highly readable and concisely illustrated treatment of the mechanical model of centrifugation for the biologist. A classic and still relevant text on the nature of transport processes as applied to biological systems. Centromeres the centromere (kentron = center; meros = part) is the region of the mitotic chromosome that participates in chromosomal movement. The mitotic spindle attaches to a specialized structure at the centromere known as the kinetochore. The motor responsible for the movement of the chromosomes toward the spindle poles during mitosis is also located at the centromere. Morphologically, centromeres are distinguished by their appearance at metaphase and anaphase as constrictions in the chromatin and by their heterochromatin staining pattern. It does not segregate properly during cell division and is rapidly lost in successive cell cycles. Centromeres occupy different positions in chromosomes, and they are useful markers. Chromosomes with centromeres near one end are called acrocentric, those with the centromere visible at or near the middle are metacentric, and those with the centromere truly at the end are telocentric. The monocentric chromosomes found in most metazoan plants and animals almost always have centromeres embedded in segments of heterochromatin. Some organisms, such as plants in the genus Luzula, have holocentric chromosomes with diffuse centromeres. In these chromosomes the spindle attaches to the centromeric heterochromatin that is distributed along the entire length of the chromosome (1). Mammalian tissue culture cells occasionally develop chromosomes with multiple centromeres (2). Dicentric chromosomes are often the result of chromosome breakage followed by fusion. A dicentric chromosome normally breaks at anaphase, when the centromeres are pulled in opposite directions. The holocentric chromosomes found in certain plants have developed as yet unknown mechanisms to avoid this problem.
The rationale of such experiments is to identify the position of the immobilized nucleic acid in the electrophoretogram that is complementary to the sequence of the probe, so that they hybridize by Watson-Crick base pairing. The conditions for hybridization, the stringency, are regulated according to the degree of homology and complementarity between the probe and the target (1, 2). Immunoblotting the original application of protein blots was for identifying a particular antigen in a protein gel pattern by probing the blot with its corresponding antibody (5). Detecting the immunocomplex is often accomplished by using a secondary probe, for example, a goat antimouse IgG conjugated to an enzyme, such as horseradish peroxidase, when a murine monoclonal antibody is used for the primary probe. The sensitivity of these assays is increased by combining immunoprecipitation prior to gel electrophoresis. Thus, a crude sample of proteins is first immunoprecipitated with relevant antibodies, and the precipitated proteins are subsequently resolved by electrophoresis, blotted, and probed with a monoclonal antibody of interest. The use of antibodies to probe colony or plaque blots is very effective in screening expression libraries (6). Lectin Blotting To identify glycoproteins, lectins can be used to probe protein blots (7). A particular case of interest is the fact that the enzyme horseradish peroxidase is itself a glycoprotein. Thus, for example, a blot can be probed with the mannose-specific lectin concanavalin A, washed and further incubated with horseradish peroxidase directly (8). The multivalent concanavalin A binds to blotted glycoproteins containing mannose and subsequently also binds to the horseradish peroxidase without the need for chemical conjugation. Ligand Blotting Blotting is usually considered for detecting antigens or nucleic acid hybrids, but it is also a very powerful way to identify all sorts of proteinprotein interactions, even interactions involving nonpeptide ligands. Thus when studying any receptor, one should consider probing protein blots with any corresponding ligands that are detectable (9). In such experiments, it is usually advisable not to boil the protein sample or subject it to disulfide bond reduction before electrophoresis. Furthermore, the blot can be incubated in solutions that promote renaturation of the immobilized proteins. Omission of methanol from the transfer buffers used in electroblotting is also helpful for retaining functional conformations of the blotted protein. Ligand blotting has been used successfully to identify peptides that bind to hormones, cytoskeletal components, neurotoxins, nucleotides, calmodulin, and even ions such as Ca2+ (9-11). The arrows indicate the position of the fusion protein containing the toxin binding site. Cell Blotting Blots have even been probed with intact cells, which has proven useful for identifying interactions between proteins and cells (12). Furthermore, bacteria can be used to probe a blot, and the interaction with a protein is detected by subsequently allowing the bacteria to grow on the surface of the blot, so that colonies are observed at the site of the immobilized protein (13). Viruses have also been used to probe blots to detect their corresponding receptor proteins. In summary, one should consider probing blots with selected ligands any time bimolecular interactions are to be analyzed. Surveying literature databases using specific conjugations, such as "ligand-blot", "calmodulin-blot" or "cell-blot," as key words usually produces results that provide the imaginative investigator with starting points from which to proceed. Blotting Blotting is a method in which a macromolecule is immobilized on a blotting matrix and subsequently probed with a detectable ligand to determine whether the macromolecule binds that specific ligand. The macromolecule can be applied to the blotting matrix directly (dot blot), or it can be derived and eluted from an electrophoretic gel (gel blot) or even from a bacterial colony or bacteriophage plaque (colony blot). After electrophoresis, the gel is dismantled from its cassette or glass plates, etc. The transfer of the resolved polynucleotides or peptides is accomplished via a procedure called "blotting", and the blotted macromolecules adsorb to the surface of the matrix while retaining their relative positions, thus creating a faithful replica of the original electrophoretic pattern. The "blot" thus produced is subsequently incubated with a ligand probe, which might be radioactive for detection via autoradiography or conjugated to an enzyme whose activity is detectable (see Blot Overlays). Extensive washing of the blot removes the excess probe from that which is specifically associated with the immobilized macromolecule and remains bound. Subsequent detection of the retained ligand in the complex formed identifies the relevant bimolecular interaction. Dot Blotting this is the simplest method of applying a sample to be tested (5, 6).
References:




